3. Start Analysis on CGAR¶
To start analyzing any of finished samples, simply click Report from the top menu to open the dialog as follows.
See also
Refer to Upload Own Variant Files for how to add new samples or check if the samples are ready.
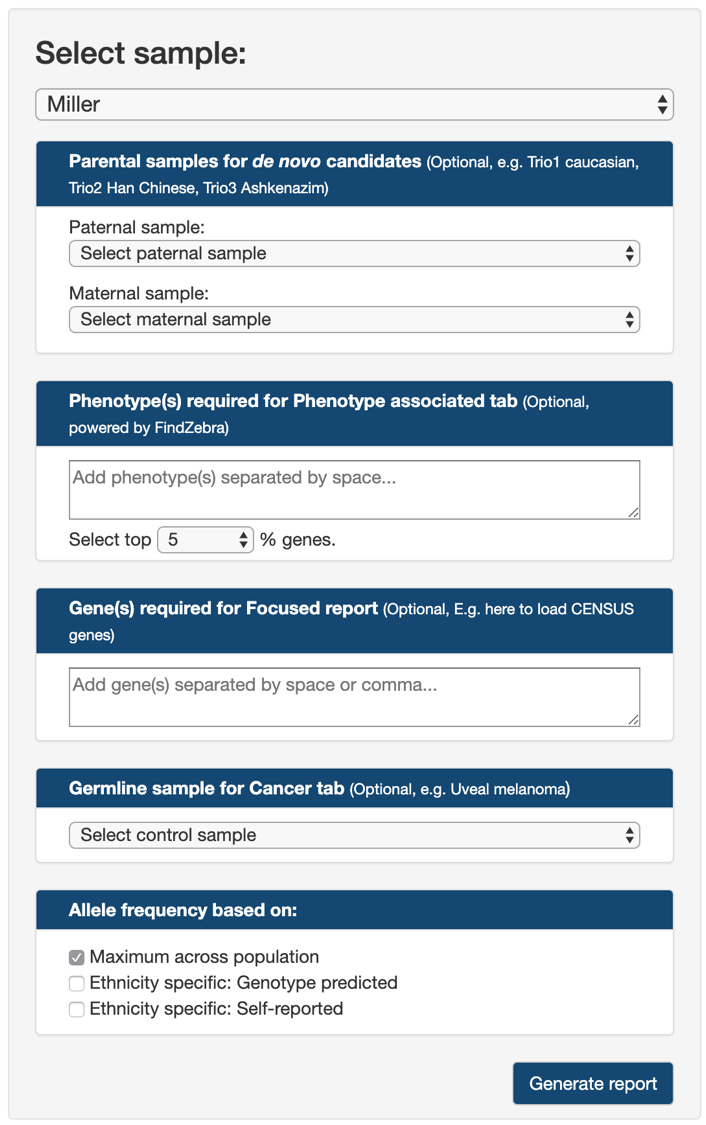
The dropdown list at the top contains analysis-ready samples for users to start work on. The CGAR public server provides a list of publicly available samples for all users who want to try out CGAR. See Publicly available samples.
If available, users can specify parental samples for the selected case in Parental samples for de novo candidates.
The dropdown lists Paternal sample and Maternal sample, both also populated with analysis-ready samples, are used to specify paternal and maternal cases, respectively.
For the de novo candidate analysis to work, both Paternal sample and Maternal sample needs to be specified.
See also
Refer to De novo variant candidates for more details on trio analysis using CGAR.
For analysis using Publicly available samples, three short-cut links are provided for each of three trios (Trio1 caucasian, Trio2 Han Chinese, and Trio3 Ashkenazim).
In cases where users are interested in variants on genes associated with specific phenotypes, yet not certain about which genes to look for, they can provide a list of keywords describing the phenotype of interest in Phenotype(s) required for Phenotype associated tab.
Users can type in as many keywords as they want, each separated by space.
CGAR will find any variants on genes associated with any of keywords.
See also
Refer to Putative phenotype-associated genes for more details on how the genes associated with the given keywords are used in CGAR.
For more focused analysis on variants in pre-defined set of genes, users can provide their genes of interest through Gene(s) required for Focused report.
CGAR will expect a list of official gene symbols (if uncertain, check the approved gene symbols from HUGO Gene Nomenclature Committee, each separated by either comma or space.
For the convenience, the list of cancer-associated genes as curated by Cancer Gene Census is provided as short-cut link.
When a matched control for cancer sample is available, specifying it as Germline sample for Cancer tab would make CGAR display only somatic variants on Cancer subsection.
Again, a short-cut for public example of uveal melanoma case (tumor and its matched blood) is provided for convenience (Uveal melanoma).
Finally, users can specify how to utilize population-specific allele frequencies in CGAR.
- Maximum across population: Default choice. Limits an allele frequency of a variant to the highest value (most common) across all populations. Results in the most strict selection of rare variants.
- Ethnicity specific: Genotype predicted: Limits an allele frequency of a variant to the value from a population of the same global ancestry as estimated from variants’ genotypes.
- Ethnicity specific: Self-reported: The same as the above, but uses the ancestry specified upon uploading the variants file. Not available if Unknown/Unspecified is chosen.
See also
Check Genotype-based prediction of ancestral composition for details on genotype-based ancestry prediction in CGAR.
After setting all the above options, click the button Generate report for CGAR to start generating report tables.
3.1. Publicly available samples¶
The following samples are readily available to all users in CGAR public server.
- Maternal trio of CHPH/Utah pedigree 1463
- Mother (NA12878) as trio1_NA12878_daughter.
- Maternal grandfather (NA12891) as trio1_NA12891_father.
- Maternal grandmother (NA12892) as trio1_NA12892_mother.
- The Han Chinese trio from Genome in a Bottle Consortium.
- The son (NA24631) as trio2_GM24631_son.
- The father (NA24694) as trio2_GM24694_father.
- The mother (NA24695) as trio2_GM24695_mother.
- The Ashkenazim trio from Genome in a Bottle Consortium.
- The son (NA24385) as trio3_NA24385_son.
- The father (NA24149) as trio3_NA24149_father.
- The mother (NA24143) as trio3_NA24143_mother.
- A pair of tumor sample with its matched blood control from uveal melanoma (MIM: 155720) case ref.
- Tumor sample as patient1_cancer_melanoma.
- Control sample as patient1_blood_melanoma.
- Artifical examples of specific phenotypes.
3.2. An Example Use Case¶
3.2.1. Method - filter variants by allele frequency¶
Let’s use an artificial sample for Miller syndrome to illustrate steps to identify variants potentially associated with phenotype.
First, select the sample Miller from the list of samples, then click Generate report to start analysis.
By default, the ClinVar tab will show 6 variants as shown below.
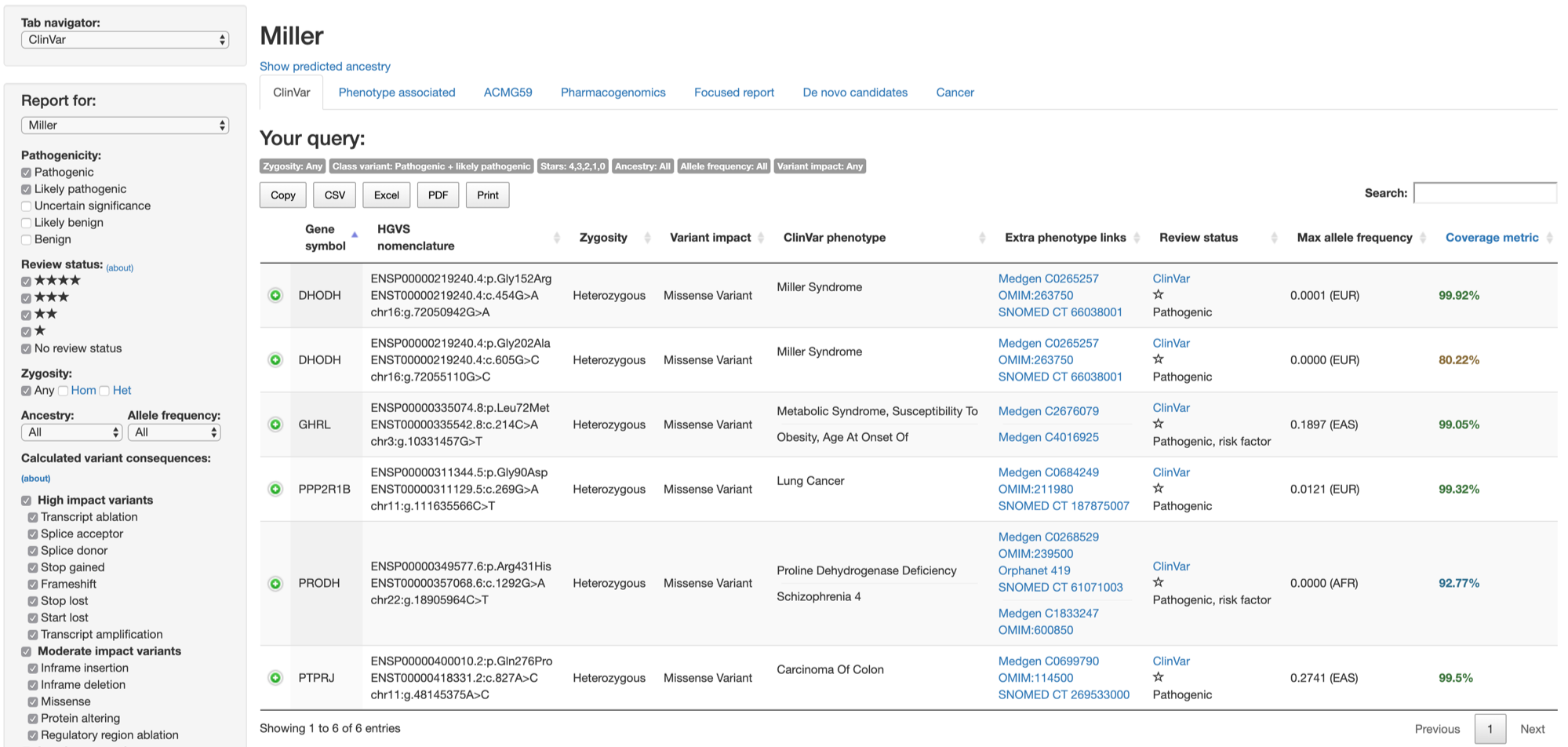
However, given the fact that the incidence of Miller syndrome is one in a million newborns (ref), it could be useful to restrict our search to vary rare variants (allele frequency less than 0.5%), then click Generate report.
(based on ACMG AMP guideline for the interpretation of sequence variants, Table 4: Criteria for benign variants, BS1 - allele frequency is greater than expected for disorder.)
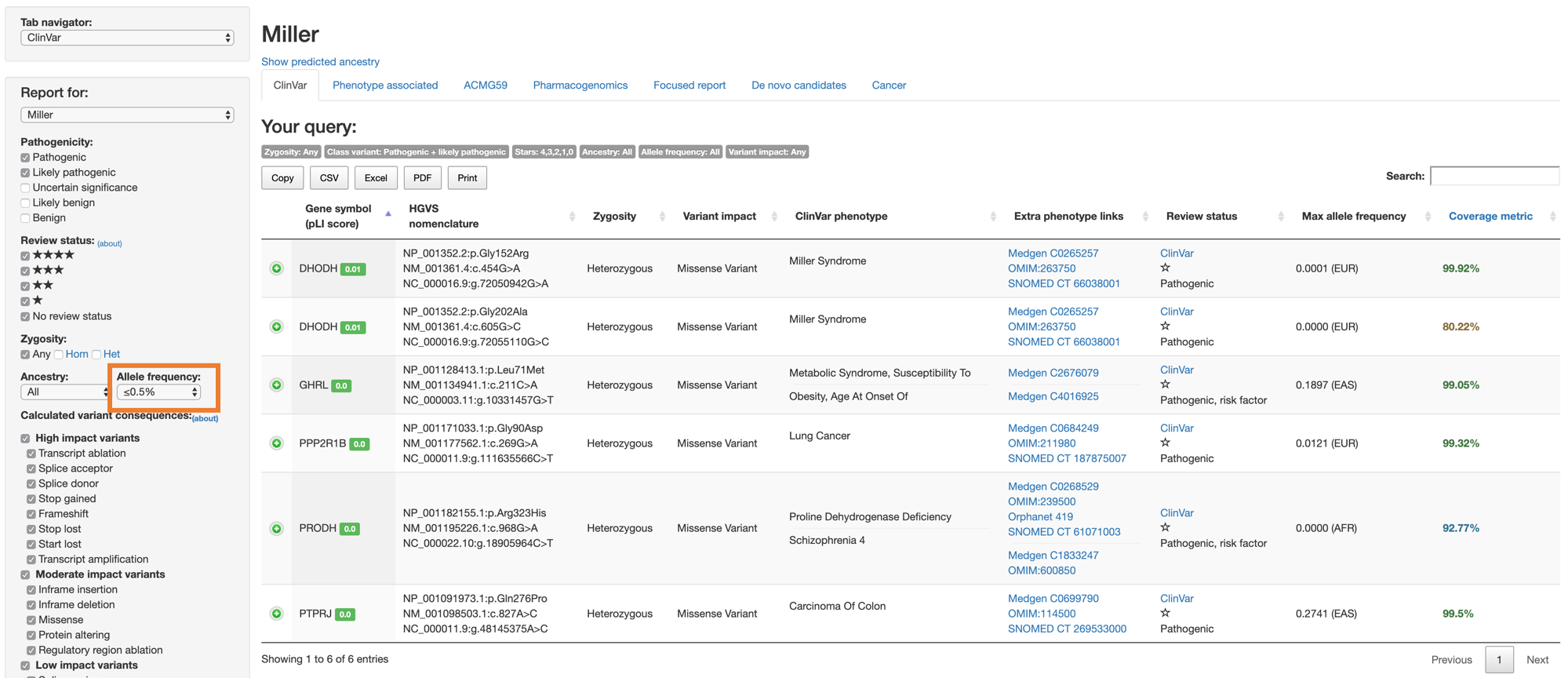
This will leave us with only 3 variants: 2 missense variants on DHODH gene and one missense variant on PRODH gene. Of these, the 2 variants on DHODH gene were the disease-assoiciated variants that were added to create artificial samples.
3.2.2. Alternative method - search variants by genes associated with phenotype¶
Alternatively, we could search for genes associated with the phenotype and identify variants on those genes, within Phenotype associated tab.
Since we know that the variants are from sample with Miller syndrome, type in Miller into the Phenotype text box.
Also, to limit our search for the most relevant genes, we select the top 1% of the genes (select 1 from the dropdown value beneath the Phenotype text box).
We also choose to see any variants that are of high or moderate impact: select both High impact variants and Moderate impact variants under the Calculated variant consequences, then click Generate report.
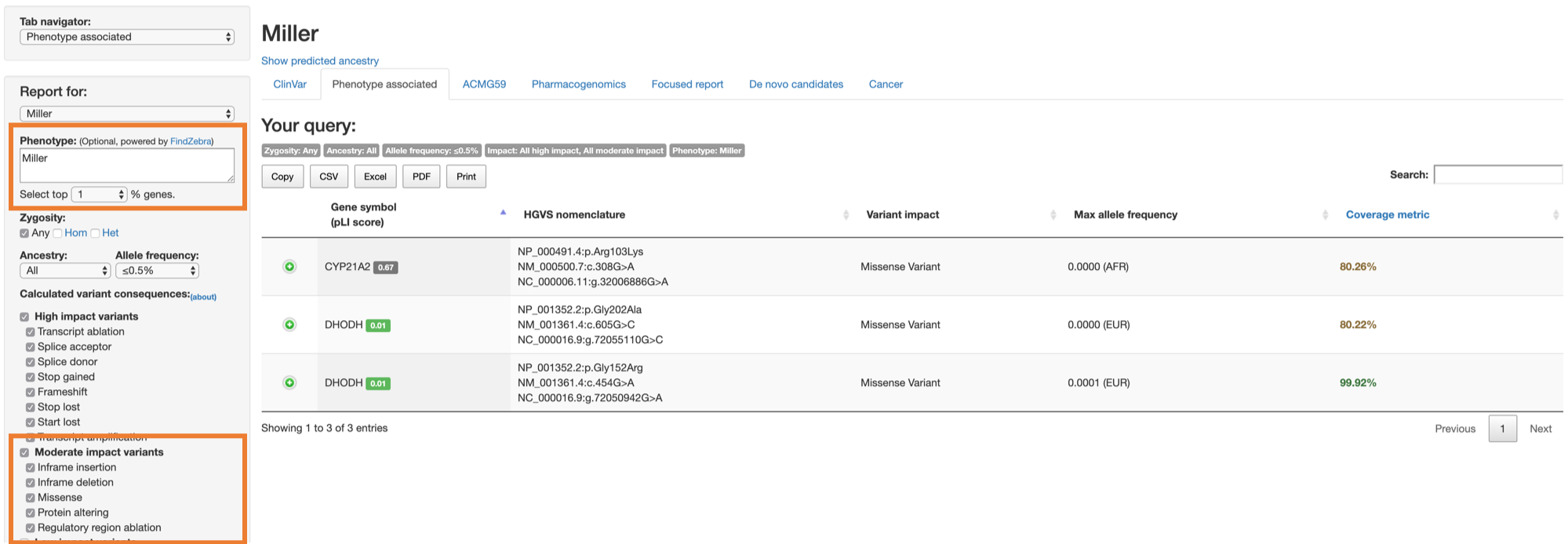
Again, we could see the 2 missense variants on DHODH gene (the same variants we found in the previous method), and another missense variant on CYP21A2 gene.
This example illustrates how CGAR can identify known disease-associated variants or rare variants in the genes associated with phenotype.