2. Upload Own Variant Files¶
Clicking Upload sample at the top menu opens a page for users to upload their own variant call file.
The files need to follow the Variant Call Format (VCF), and only one file can be uploaded at a time.
However, VCF files containing more than one individual or sample are accepted.
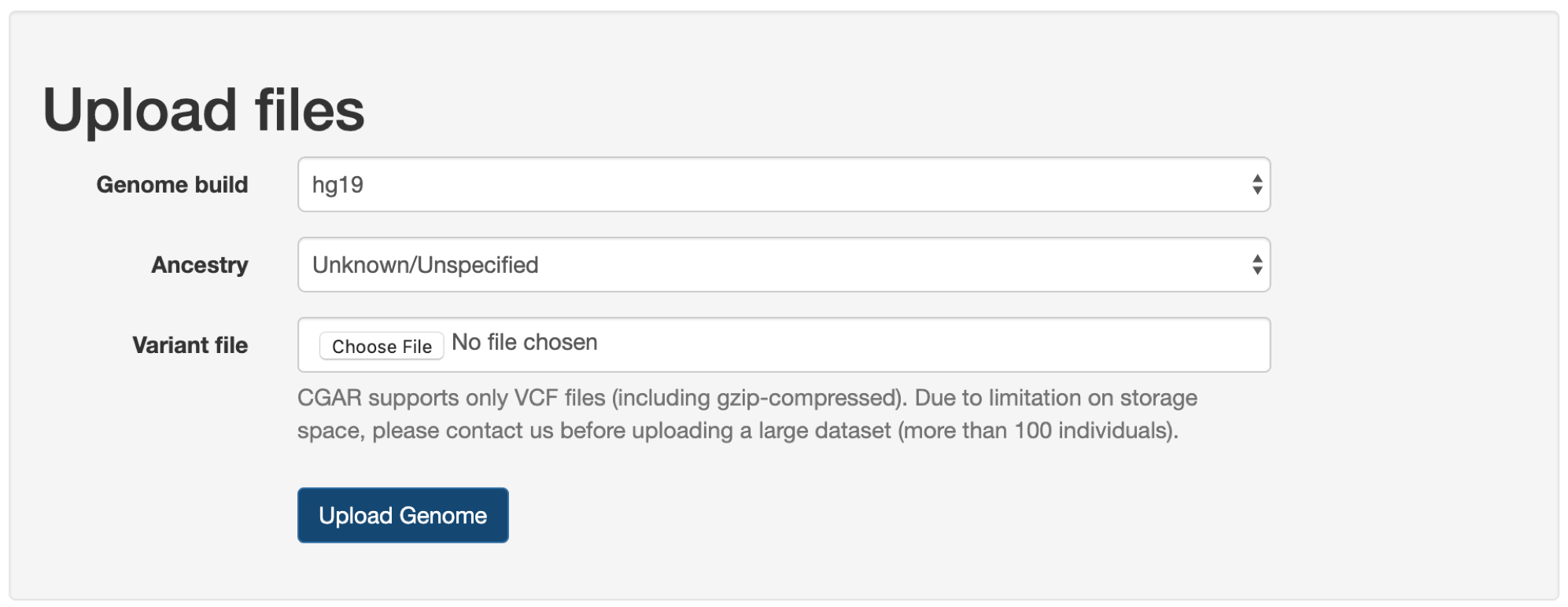
First, users need to select correct version of human reference genome assembly (Genome build): hg19 or hg38.
If uncertain, it is recommended to check the header lines within the file (lines starting with #).
To do so, you can run the following command in a terminal:
# if the file is gzip-compressed
$ zcat /path/to/variant/file.vcf.gz | head -n <arbitrary number of lines to view>
# if the file is not compressed
$ head -n <arbitrary number of lines to view> /path/to/variant/file.vcf
##fileformat=VCFv(version number)
...
Then, look for a line starting with ##reference=.
This line usually contains file name for referece genome fasta file, which could provide a hint of reference genome assembly version.
For example, if the filename contained b37, hg19, or human_g1k_v37, it’s most likely that you can choose hg19.
Or, find lines starting with ##contig=, which list the set of chromosomes with their names and lengths.
From Human Assembly Data in Genome Reference Consortium, you can find chromosome lengths in different versions of assembly, and match with values found in your VCF file.
Next, the global ancestry (Ancestry) for the individual or sample to be analyzed, if known, is recommended to be set.
The same five continental groups as in the 1000 Genomes Project are used: African, East Asian, European, South Asian, and American.
If unknown or uncertain, it can be left as Unknown/Unspecified, and CGAR will estimate the global ancestry from variant file.
In the Variant file, local variant file can be selected for uploading to CGAR.
Only files ending with the extension of .vcf or .vcf.gz are accepted.
Note
Due to limitation on storage space, please contact us before uploading a file containing large number of individuals (more than 100 individuals).
Finally, when user clicks on the Upload Genome button (at the bottom of dialog), the variant call file gets transferred to the server and placed on a queue for annotation and analysis.
The required amount of time to finish the process varies, depending on the number of variants in the file.
Under normal circumstances, a file of whole genome containing 3 to 4 million variants gets processed in one hour.
Users will receive a notification email each time a file is done processed and available for analysis.
2.1. Browse and track previous files¶
The bottom of the page displays a table listing all variant files uploaded so far (including the one you have just uploaded).
In this table, users can see:
- The name of sample(s) in the variant file -
Genome label. - The name of the variant file -
Genome file name. - The version of human reference genome assembly -
Genome build. - The global ancestry specified at the time of uploading -
Genome ancestry. - The version of annotation -
Annotation. - The time and date when the file was first uploaded or done processing -
Uploaded.
When the file was just uploaded, but not yet finished processed and ready for analysis, it would first appear in the table with none as Annotation.
Also, the Genome label would simply show the number of samples within the file, e.g., (3 sample(s)) or (1 sample(s)).
Later, the table will be updated with as many samples as was in the file, with each sample as separate row carrying their own identifier (specified in original VCF file) as Genome label.
At this point, all samples will have the current version of annotation in Annotation, and Uploaded will reflect the date and time when the processing was finished.