1. Introduction¶
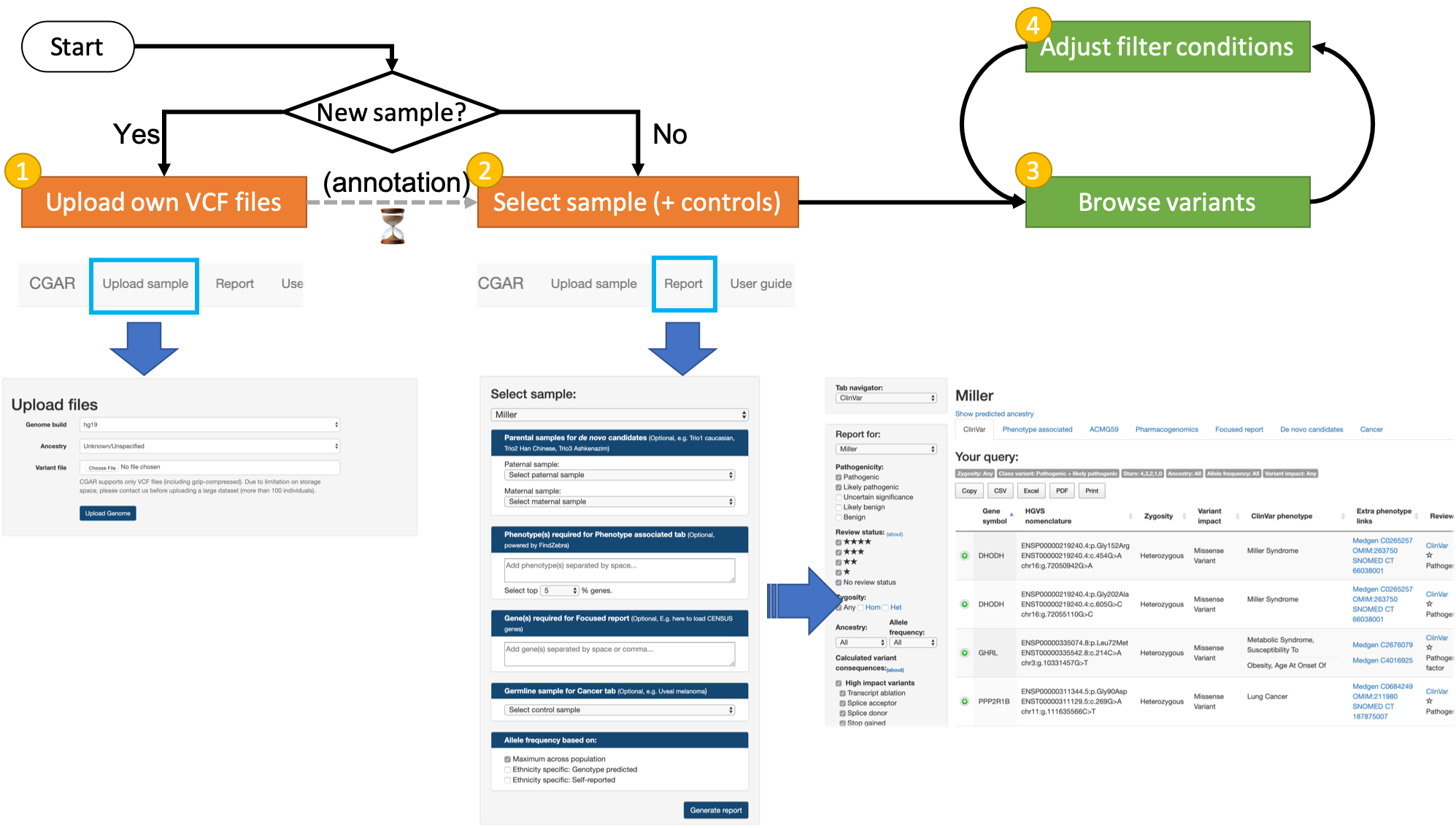
Clinical Genome & Ancestry Report (CGAR) is an interactive web application to dynamically filter and organize clinically implicated variants from variant call files in VCF. Variants are annotated with latest information from diverse sources and can be searched by simple yet comprehensive filtering options, with various links to external resources.
CGAR organizes variants according to the following categories:
- Reported disease-associated variants from HGMD® (license required) and ClinVar,
- Variants in putative disease-associated genes,
- Secondary findings recommended for reporting,
- Variants with pharmacogenomic consequences,
- Variants on user-defined genes,
- De novo variant candidates for trio (if parental samples are specified),
- Germline or somatic (if matched control sample is specified) variants implicated in cancer risk, diagnosis, treatment and prognosis.
Note
Some sections of CGAR report require specific license. Only registered users with proper license are allowed to access them.
For each category, variants selected by a set of configurable criteria are presented in intuitive and interactive web interface where users can browse and identify clinically implicated variants from thousands (or millions) of variants from whole exome (or genome) sequencing data.
1.1. CGAR public server¶
CGAR is freely available at https://tom.tch.harvard.edu/apps/cgar/.
Any interested users can use the full functionality of CGAR without registration. However, the workspace of unregistered users are shared with each other, implying that they can access the same set of samples. In other words, samples uploaded by an user A can be viewed by another user B.
Users who need private workspace are encouraged to register or to set up CGAR locally. Please contact us to inquiry about registration.
Note
Check Running CGAR locally for guidelines to set up CGAR locally.